Medme poleca





Mukopolisacharydoza
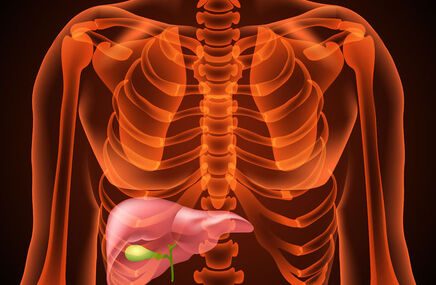
Mukopolisacharydoza występuje zaledwie raz na 100 tysięcy urodzeń. Przyczyną choroby jest wada metabolizmu, która polega na gromadzeniu się w organizmie mukopolisacharydów - części składowych tkanki łącznej. Składają się one z łańcuchów cząsteczek cukrów i podlegają rozkładowi dzięki działaniu określonej grupy enzymów. Brak lub wada jednego z tych enzymów powoduje zaburzenie procesu rozpadu, a w konsekwencji odkładanie się jego produktów i uszkodzenie komórek i narządów ciała. Prowadzi to do ich niewydolności, a co za tym idzie wyniszczenia niemal całego organizmu chorego.
Reklama
Choroba mukopolisacharydoza
Badania nad mukopolisacharydozą wykazały, że przyczyną braku lub wad enzymów jest defekt materiału genetycznego. W zależności od tego, którego enzymu brakuje, choroba może występować pod różnymi postaciami (typ I, II, III, IV, V i VI). Zdiagnozowanie choroby jest możliwe jeszcze w okresie prenatalnym.
Reklama
Mukopolisacharydoza - objawy
Jednym z pierwszych objawów mukopolisacharydozy jest przepuklina pępkowa i pachwinowa. Wcześnie występują także polipy błony śluzowej oraz problemy ze słuchem. W późniejszych etapach choroby pojawiają się zaburzenia ruchu połączone z usztywnieniem stawów, zmętnienie rogówki oraz biegunki.
Reklama
Mukopolisacharydoza typu 1
Mukopolisacharydoza typu 1 jest zwana także zespołem Hurler. To najpoważniejsza postać mukopolisacharydozy, która cechuje się ciężkim upośledzeniem umysłowym i fizycznym. Śmierć chorego następuje najpóźniej około 10. roku życia.
Reklama
Mukopolisacharydoza typ 2
Mukopolisacharydoza typu 2 jest nazywana także zespołem Huntera. Od innych typów mukopolisacharydozy różni się odmiennym sposobem dziedziczenia. Jest ono sprzężone z chromosomem X, co sprawia, że choroba dotyka zwłaszcza mężczyzn. Wyróżniane są dwie postacie zespołu Huntera. Typ A przejawia się w deformacji kręgosłupa i upośledzeniu umysłowym. Do objawów zalicza się także m.in. pogrubienie rysów twarzy, niski wzrost, powiększenie wątroby i śledziony oraz typowe dla mukopolisacharydozy przepukliny pępkowe i pachwinowe.
W typie B zmiany kostne przebiegają łagodniej, a upośledzenie jest umiarkowane lub nie występuje w ogóle. Charakterystyczne dla obu typów zespołu Huntera są zmiany skórne o charakterze małych grudek umiejscowionych na ramionach, łopatkach i plecach. Objawy mukopolisacharydozy typu 2 pojawiają się zwykle między 2 a 3 rokiem życia dziecka. Okres przeżycia osoby chorującej na zespół Huntera typu A to około 20 lat. Pacjenci z typem B żyją nawet do dwóch razy dłużej.
Reklama
Mukopolisacharydoza typ 3
O mukopolisacharydozie typu 3 mówi się także jako o zespole Sanfilippo. Jego objawy ujawniają się zwykle po trzecim roku życia dziecka. Wówczas zaczyna się ono cofać w rozwoju psychoruchowym. Początkowe objawy choroby nie są charakterystyczne, a zmiany w zachowaniu i przeżywaniu emocji mogą sugerować autyzm. Dziecko zaczyna mieć trudności z koncentracją i pamięcią, w końcu przestaje nawet prawidłowo mówić. Po pewnym czasie pojawia się niepełnosprawność ruchowa. Maluch niepewnie stoi na nogach i często traci równowagę, by ostatecznie utracić zdolność chodzenia. Finalnie dochodzi również do zmian rysów twarzy, m.in. pogrubienia warg i poszerzenia nosa. Rokowania są bardzo złe – chory zwykle nie dożywa pełnoletności i najczęściej umiera w wieku 13 lub 14 lat.
Reklama
Mukopolisacharydoza - leczenie
Mukopolisacharydozy nie da się wyleczyć, ponieważ metody naprawy uszkodzonego materiału genetycznego nie są jeszcze znane. Objawy choroby można jedynie złagodzić. W przypadku niektórych typów mukopolisacharydozy złagodzenie schorzenia można osiągnąć poprzez przeszczep szpiku kostnego, jednak zabieg ten wiąże się z dużym ryzykiem.