Choroba Pompego zaliczana jest do grupy chorób genetycznych związanych z pośrednim upośledzeniem funkcji organelli komórkowych zwanych lizosomami (dlatego też o chorobie Pompego mówi się, iż należy do grupy chorób spichrzeniowych). Glikogenoza II jest rzadkim schorzeniem – występuje ze średnią częstością populacyjną 1 na 40 000 żywych urodzeń – jednakże, ze względu na charakter kliniczny jednostki dane szacunkowe mogą być obarczone znacznym błędem.
W przeciwieństwie do wielu innych chorób genetycznych, w tym spichrzeniowych lizosomów, glikogenowa II może objawić się w różnym wieku pacjenta z różnym nasileniem, dlatego też wprowadzono stosowny podział kliniczny choroby:
- postać noworodkową,
- postać wieku młodzieżowego
- postać u osób dorosłych.
Nieleczona choroba powoduje zgon jeszcze w okresie wczesnego dzieciństwa.
Choroba Pompego - przyczyny
Bezpośrednią przyczyną wystąpienia choroby Pompego jest mutacja genu kodującego enzym zwany alfa-glikozydazą (kwaśną maltazą, GAA). Mutacje (około 40 różnych) mają charakter zmian punktowych o dziedziczeniu autosomalnym, recesywnym. Następstwem mutacji jest dysfunkcja lub znacznie obniżony poziom enzymu – w przypadku postaci noworodkowej zdarza się, iż aktywność GAA nie przekracza 1%, podczas gdy w przypadku pacjentów dorosłych ten współczynnik może dochodzić nawet do 45%.
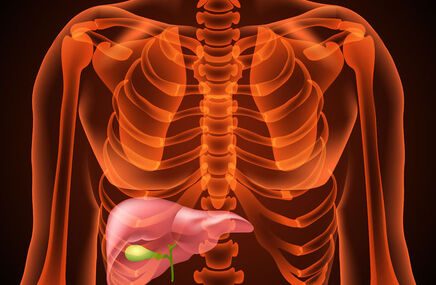
Enzym ten należy do grupy enzymów lizosomalnych odpowiedzialnych za hydrolizę (czyli rozkład enzymatyczny) glikogenu. W efekcie zablokowania szlaków metabolicznych lub ich upośledzenia, poprzez niewydolną pracę, dochodzi do gromadzenia się stale przybywającego glikogenu w lizosomie. Powstające agregaty glikogenu powodują w następstwie uszkodzenie lizosomu i całej komórki. Najbardziej narażone są:
- komórki wątroby,
- komórki mięśniowe,
- komórki serca,
- komórki glejowe
- niektóre komórki układu nerwowego (jąder ruchowych pnia mózgu i komórki rogów przednich rdzenia kręgowego).
W zależności od zaistniałej zmiany enzymatycznej procesy agregacji glikogenu mogą trwać latami lub dać pierwsze objawy już w okresie noworodkowym. Niestety z obserwacji klinicznych wynika, iż im wcześniej choroba manifestuje się u pacjenta, tym objawy kliniczne są znacznie silniejsze i szybciej postępujące.
Reklama
Choroba Pompego - objawy
Do głównych objawów choroby Pompego zalicza się;
- postępujące osłabienie mięśni (zwłaszcza mięśni szkieletowych oraz mięśnia sercowego). Następstwem problemów z układem mięśniowym jest znacznie ograniczone poruszania się, problemy z oddychaniem (objawy bezdechu sennego w momencie ucisku na klatkę piersiową osoby śpiącej), zaburzenia rytmu serca, problemy z przyjmowaniem pokarmu – u niemowląt może to prowadzić do braku odruchu ssania;
- postępujące osłabienie mięśni prowadzi do wystąpienia wad postawy np. do skolioz;
- poranne bóle głowy;
- zawroty głowy;
- łatwość męczenia się;
- powiększony miesień sercowy;
- powiększona wątroba;
- powiększony język.
Choroba Pompego - diagnoza
Rozpoznanie choroby Pompego może nastąpić zarówno we wczesnym okresie noworodkowym, jak w wieku dorosłym. Większość objawów obserwowanych w stanach łagodnych i ostrych choroby nie zawsze daje możliwość jasnej diagnozy klinicznej.
W chwili obecnej w celu potwierdzenia diagnozy stosuje się testy biochemiczne i monitoring pracy serca.
- W przypadku testów biochemicznych dokonuje się oceny poziomu i aktywności GAA. W tym celu przeprowadza się biopsję skóry i/lub mięśnia poprzecznie prążkowanego.
- Uzupełnieniem testów biochemicznych jest również określenie poziomu stężenia we krwi pacjenta enzymów wątrobowych oraz kinazy kreatyninowej (w przypadku choroby Pompego stężenie ich jest podwyższone).
- W diagnostyce stosuje się również badania mające na celu ocenę pracy oraz stanu serca - elektromiografia, RTG klatki piersiowej, EKG, echo serca.
- W przypadku par, w których oboje partnerzy mają obciążony wywiad genetyczny, istnieje możliwość przeprowadzenia badań prenatalnych. Podobnie jak u pacjentów w okresie postnatalnym również w tym przypadku wykonuje się pomiar ilość i aktywności enzymatycznej GAA. Materiał do badań stanowią komórki płodu pobrane metodą amniopunkcji z płynu owodniowego.
- Istnieje możliwość wykonania badań genetycznych, jednakże ze względu na bardzo niską częstość choroby oraz jej heterogenną etiologię, analizy genetyczne stosowane są w ośrodkach naukowych i eksperymentalnych.
Głównym czynnikiem ryzyka jest obecność mutacji u jednego lub obojga rodziców. Jak wspomniano mutacja powodująca wystąpienie objawów klinicznych ma charakter zmiany autosomalnej, recesywnej tzn. dziedziczy się w sposób niezależny od płci dziecka i do wystąpienia choroby konieczna jest obecność zmutowanych obu kopii genu od ojca i od matki.
W przypadku obecności tylko jednej nieprawidłowej kopii genu nie dochodzi do wystąpienia jakichkolwiek zaburzeń, a osoba obciążona taką zmianą jest określana nosicielem mutacji. Szacunkowe ryzyko wystąpienia choroby u dziecka (kiedy wywiad rodzinny nie wykazuje żadnych obciążeń) wynosi około 0,1%. Jednakże w przypadku par, w rodzinach których występowała choroba (zwłaszcza wśród krewnych I i II stopnia), ryzyko wystąpienia choroby jest znacznie wyższe – może ono wynosić nawet 25%, jeżeli oboje rodzice są nosicielami mutacji. W takiej sytuacji istnieje również ryzyko, iż połowa dzieci takiej pary będzie również bezobjawowymi nosicielami mutacji.
Ze względu na etiologię choroby, w chwili obecnej nie istnieją żadne skuteczne sposoby zapobiegania.
Reklama
Choroba Pompego - leczenie
W przypadku chorób o podłożu genetycznym nie jest możliwe wprowadzenie skutecznego leczenia przyczynowego, a jedynie leczenie doraźne, którego celem jest niwelowanie objawów klinicznych.
W przypadku choroby Pompego leczenie ma na celu wyrównanie aktywności i poziomu brakującego enzymu – w chwili obecnej prowadzone są terapie eksperymentalne.
Nieleczona choroba Pompego jest niestety wyrokiem dla pacjenta. Podjęcie leczenia choroby również nie powoduje jest zahamowania, a jedynie spowolnienie postępowania objawów klinicznych. W przypadku noworodków lub bardzo małych dzieci już na wczesnym etapie rozpoznania konieczna jest interwencja (wielu pacjentów ma problemy z połykaniem pokarmu, dlatego też stosowane są metody karmienia przez sondy).
W przypadku starszych pacjentów, u których główne objawy choroby skupione są na układzie mięśniowym, konieczne jest podjęcie właściwej terapii treningowej. Niestety nasilające się problemy mogą zmusić pacjenta do korzystania z wózka inwalidzkiego i aparatury wspomagającej oddychanie. Osłabienie mięśni, w tym mięśni oddechowych, ogranicza możliwości chorych w wykonywaniu pewnych czynności jak również uniemożliwia wzmożony wysiłek fizyczny.
- Sprawdź czym jest: Choroba Fabry'ego
Jakie badania należy wykonać, aby wykluczyć nosicielstwo mutacji?
Mutacje powodujące wystąpienia choroby Pompego mają charter zmian recesywnych, co oznacza, iż do wystąpienia choroby konieczna jest obecność mutacji w obu kopiach genów. W przypadku nosicielstwa tylko jedna kopia ulega mutacji podczas gdy druga koduje prawidłowo funkcjonujący enzym. Nosiciele nie wykazują jakichkolwiek objawów choroby. Jedynym badaniem dającym 99,9% pewność co do obecności mutacji jest badanie genetyczne.